

Les produits pharmaceutiques sont un pilier crucial des systèmes de santé modernes : ils constituent le troisième poste des dépenses de santé après les soins stationnaires et ambulatoires. Selon les estimations, la valeur du marché pharmaceutique mondial devrait dépasser 1500 milliards de dollars en 2023[1]. Pour donner une idée du contexte, les dépenses mondiales de santé étaient au total estimées à 8300 milliards de dollars en 2018[2]. La recherche et développement (R&D) des pharmas n’est pas non plus en reste : entre 2010 et 2020, l’Agence américaine des produits alimentaires et des médicaments (FDA) a approuvé en moyenne 43 nouveaux médicaments par an[3]. Ce chiffre est de 31 en Suisse pour la période 2002–2020 (voir illustration 1). Bien qu’elle reflète le souhait général des patients, du monde politique et des producteurs de faciliter l’accès à des thérapies potentiellement révolutionnaires, cette succession rapide de nouveaux produits peut représenter un défi pour les gouvernements et les fabricants.
Ill. 1. Approbation de nouveaux médicaments en Suisse (2002–2020)
Source : Swissmedic / La Vie économique
Un point crucial concerne l’incertitude souvent importante entourant le bénéfice clinique d’un nouveau médicament lors de son entrée sur le marché. Les autorités de régulation des médicaments du monde entier sont mises au défi d’accélérer l’accès au marché. Bon nombre d’entre elles ont établi des procédures accélérées pour les traitements prometteurs répondant à d’importants besoins non couverts. C’est particulièrement le cas des nouveaux médicaments contre le cancer. Ceux-ci se voient souvent accorder une autorisation de commercialisation à un stade plus précoce de leur développement ou sur la base de critères de substitution – par exemple des mesures indirectes de l’effet d’un traitement censées prédire des issues cliniques intéressantes comme la survie ou l’amélioration de la qualité de vie. D’autres peuvent aussi se baser sur des preuves issues d’études non randomisées. Et même dans les cas où les produits ne sont pas homologués en procédure rapide, les preuves tirées d’essais randomisés contrôlés (qui excluent souvent les personnes très âgées, très jeunes et celles souffrant de plusieurs pathologies) peuvent être difficiles à généraliser à des populations plus vastes de patients.
Deux problèmes majeurs
Deux problèmes majeurs se posent aux payeurs publics et aux agences d’évaluation des technologies de la santé qui examinent les nouveaux traitements. Le premier est de déterminer si une nouvelle thérapie constitue un progrès significatif par rapport aux alternatives existantes, et si oui, de mesurer et d’estimer ensuite la valeur de cette différence. Les incertitudes résultant de données issues d’essais cliniques immatures ou limités créent de nouvelles zones de doute lorsqu’il s’agit de déterminer l’efficacité clinique comparative et, par extension, l’efficacité des coûts et l’impact budgétaire potentiel. Ajoutées aux prix de lancement toujours plus élevés des nouveaux médicaments, ces incertitudes mettent au défi la capacité des payeurs à prendre des décisions sûres en matière de prise en charge et de prix, et à estimer les dépenses futures.
Accélérer l’entrée de produits sur le marché peut également être à double tranchant pour les pharmas. Dans de nombreux pays, il n’existe pas de marché significatif pour les nouveaux médicaments tant que ceux-ci n’ont pas été listés dans des catalogues de soins nationaux, régionaux ou institutionnels[4]. Si la preuve de leur efficacité est limitée, les fabricants peuvent connaître d’importantes difficultés à démontrer aux payeurs et aux agences d’évaluation des technologies de la santé le bénéfice de leurs produits et, par conséquent, à obtenir leur inclusion ou leur prise en charge dans un catalogue de soins à un prix qu’ils jugent acceptable.
Accords entre payeurs et fabricants…
Comment les pays s’efforcent-ils de gérer ces défis ? Une approche recourt à des « contrats d’accès au marché » (CAM, voir illustration 2)[5]. Il s’agit d’accords négociés entre payeurs et entreprises pharmaceutiques qui permettent de prendre en charge de nouveaux médicaments tout en gérant l’incertitude quant à leur impact financier ou leur performance clinique. La plupart se concentrent cependant souvent sur la gestion des risques budgétaires et ne réduisent pas les incertitudes entourant le bénéfice clinique des traitements. Les CAM financiers sont utilisés dans au moins deux tiers des États membres de l’Organisation de coopération et de développement économiques (OCDE) et de l’Union européenne (UE).
Ill. 2. Politiques et approches de l’incertitude dans l’OCDE et les pays de l’UE
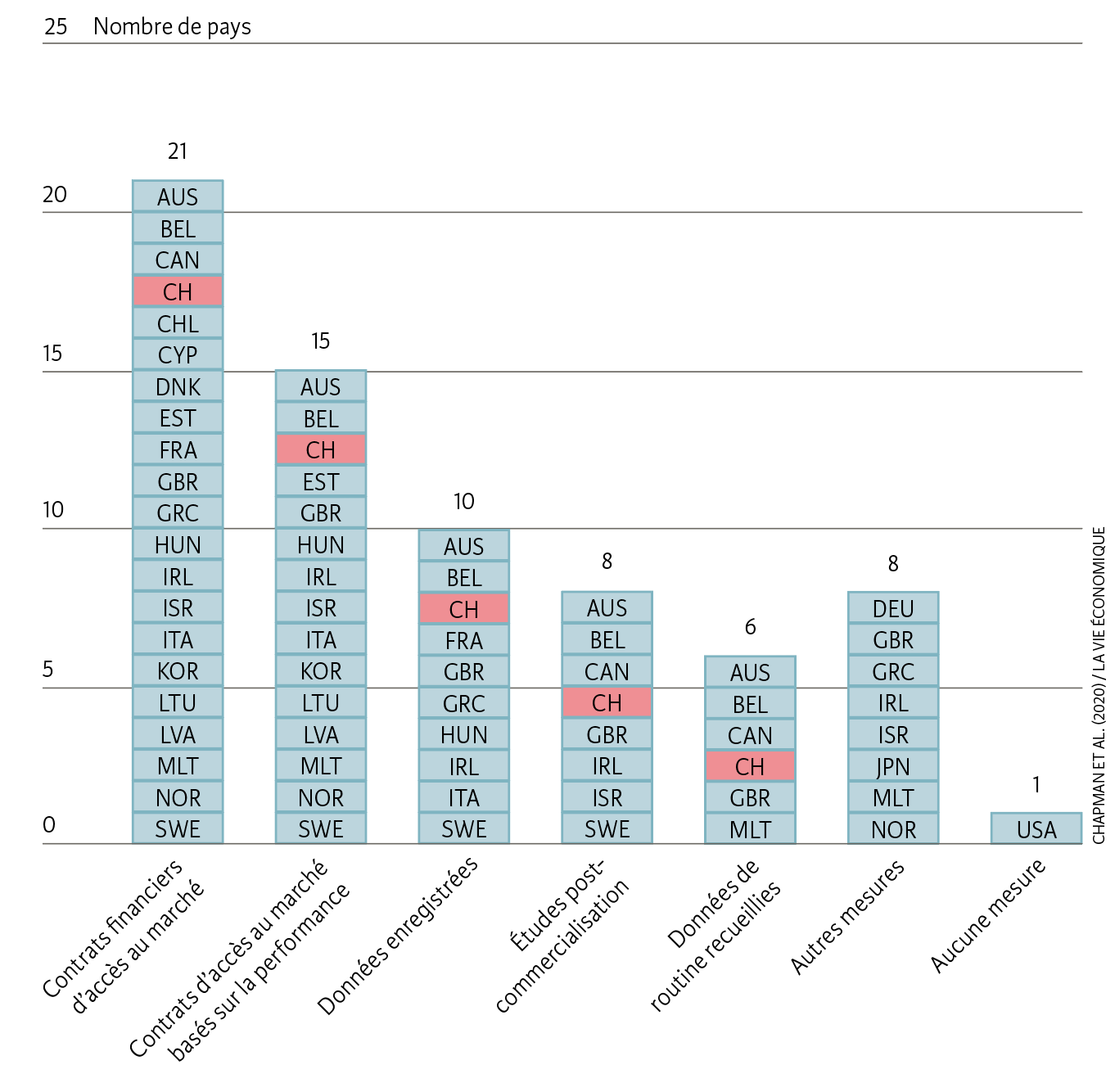
Remarque : le graphique a été établi sur la base des réponses de 24 pays à une enquête de l’OCDE concernant les défis en matière d’accès aux médicaments oncologiques. Les réponses multiples étaient possibles.
Source : Chapman et al. (2020) / La Vie économique
Plusieurs pays recourent également à des CAM « basés sur la performance ». Moins courants, ces accords lient la prise en charge, les paiements aux entreprises ou les remises versées par les entreprises à la performance des produits[6]. Un modèle de CAM basé sur la performance est le paiement « au résultat » évalué au niveau du patient : cette formule peut améliorer l’efficacité des coûts et aider les payeurs à gérer l’impact budgétaire en stipulant que les fabricants ne seront payés que pour les traitements auxquels les patients répondent. Un autre modèle de CAM basé sur la performance est la prise en charge au niveau de la population conditionnée sur le recueil de données probantes : cette option est utilisée pour gérer l’incertitude quant à l’efficacité comparative ou l’efficacité des coûts.
L’Italie, l’Estonie, la Corée du Sud et le Royaume-Uni font partie des pays à avoir introduit des CAM basés sur la performance. La première nommée est de loin celle qui a accumulé le plus d’expérience à ce jour. Il est malheureusement difficile d’estimer le succès de ce modèle, car peu de pays ont entrepris des évaluations formelles.
Le caractère confidentiel de la plupart des CAM ne facilite pas la réalisation d’évaluations indépendantes. Les exemples limités recueillis suggèrent toutefois que les accords concernant une prise en charge conditionnée sur le recueil de données probantes n’ont guère réussi à réduire l’incertitude entourant la performance des médicaments et que l’usage de ce type de CAM décline. À l’opposé, les contrats de paiement au résultat continuent d’être très largement utilisés, mais ils ne produisent pas toujours de preuves quant à la performance des produits, les données utilisées pour déclencher les paiements n’étant pas toujours agrégées et analysées à cette fin. En outre, la surcharge administrative liée à la mise en œuvre des CAM et à la collecte et à l’analyse des données concernant la performance des médicaments peut être substantielle. Une attention accrue à la conception des CAM et un surcroît d’approches harmonisées entre pays pourrait en renforcer l’utilité.
… et données tirées du terrain
D’autres approches comprennent le recours aux données dites « réelles », recueillies dans la pratique clinique ordinaire (dossiers électroniques compris). Ce type de stratégie est utilisé par de nombreux pays de l’OCDE et de l’UE pour compléter les données issues d’essais cliniques et destinées à résoudre les incertitudes relatives à la performance clinique et à l’efficacité des coûts[7]. L’Italie utilise par exemple des registres virtuels à l’échelle nationale pour surveiller l’usage correct des médicaments dans la pratique clinique et le respect des conditions financières des CAM : dans ce cadre, les données servent également à résoudre les incertitudes concernant la longueur des traitements ou la durée de leurs effets[8].
Quelques pays s’en remettent aussi aux études post-commercialisation pour résoudre l’incertitude au moment de l’autorisation de mise sur le marché. En Allemagne, une législation récente[9] permet par exemple à l’agence d’évaluation des technologies de la santé de demander à une société pharmaceutique de collecter les données de pratique clinique en vue d’en évaluer le bénéfice. Cette possibilité est toutefois limitée aux médicaments sous approbation conditionnelle ou exceptionnelle et aux médicaments « orphelins ».
Collaboration et engagement
Dans une enquête récente[10] consacrée aux défis particuliers rencontrés par les pays de l’OCDE en matière d’accès aux médicaments oncologiques, tous les pays participants voyaient un intérêt à une collaboration internationale accrue pour résoudre les incertitudes cliniques et/ou économiques concernant les nouveaux médicaments. Et ce, même si certains ont émis des réserves résiduelles quant à la faisabilité d’un partage des informations et d’une généralisation de données nationales à l’ensemble des populations et des systèmes de santé.
Plusieurs collaborations de ce type se sont déjà mises en place[11]. L’initiative BeNeLuxA est l’une des plus abouties : visant à soutenir un accès durable aux traitements novateurs et lancée en 2015 par la Belgique et les Pays-Bas, elle s’est élargie depuis au Luxembourg, à l’Autriche et à l’Irlande. Le projet implique la coopération en matière de prospective, d’évaluation des technologies de la santé et de négociations sur les prix et les remboursements. BeNeLuxA a récemment conclu avec succès sa première négociation conjointe sur les prix. Deux autres collaborations de pays européens – le Forum nordique sur les médicaments et l’Initiative balte sur l’approvisionnement en médicaments – ont entrepris avec succès des processus conjoints d’appel d’offres pour des médicaments et des vaccins. Toutes ces initiatives évoluent cependant constamment et doivent encore exploiter tout leur potentiel, au fur et à mesure que les pays gagnent en expérience collaborative.
Quelles autres mesures pourraient être prises ? Certains programmes d’essais cliniques sont souvent conçus avant tout en fonction des besoins des régulateurs. Les agences d’évaluation des technologies de la santé qui essaient de déterminer la valeur d’un traitement auront ainsi tendance à rechercher des preuves des améliorations de la qualité de vie et d’autres résultats rapportés par les patients et à y accorder du poids, ce qui ne peut pas être habituellement exigé par les régulateurs. Une collaboration accrue et plus précoce entre les autorités de régulation des médicaments, les agences d’évaluation des technologies de la santé et les payeurs, ainsi qu’entre ces entités et les sociétés pharmaceutiques, pourrait aider ces dernières à concentrer leurs programmes de R&D sur la mise au point de preuves pertinentes à la fois pour les régulateurs et pour les agences d’évaluation des technologies de la santé. Une telle démarche pourrait renforcer la probabilité de décisions éclairées et d’issues positives pour toutes les parties prenantes.

- Miglierini (2019).
- OMS (2020).
- OCDE (2021).
- Un catalogue de soins est une liste de médicaments couverts par une police particulière d’assurance-maladie ou approuvés pour ordonnance dans un système de santé ou un hôpital spécifique.
- Pour une vision critique des CAM, voir également l’article de Thomas Christen et Jörg Indermitte (OFSP) dans ce numéro.
- Wenzl et Chapman (2019).
- OCDE (2020)
- Montilla et al. (2015).
- Loi allemande pour plus sécurité dans l’approvisionnement en médicaments, § 35a (3b).
- Chapman et al. (2020).
- Vogler et al. (2021).








